- 移动端


研载生物科技(上海)有限公司
8 年
手机商铺
商家活跃:
产品热度:
- NaN
- 0.5
- 1.5
- 0.5
- 3.5

公司新闻/正文
多重热点联合:外泌体+circRNA+铁死亡,1区15分+文章轻松到手!
2658 人阅读发布时间:2022-08-15 11:24
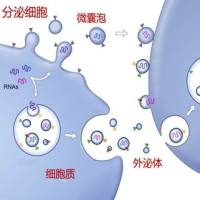
肺腺癌 LUAD 是肺癌最普遍的亚型,尽管 EGFR、KRAS、ALK 和 TP53 基因突变,以及 m6A-RNA 甲基化失调是 LUAD 的主要诱因,但最近研究发现抗铁死亡是与 LUAD 的起始和发展相关的另一种非突变机制。先前研究集中在细胞内抗氧化系统,细胞外系统是否使 LUAD 对铁死亡脱敏仍不清楚。作为细胞外系统中信号传导的主要调节剂,外泌体 EV 是否参与 LUAD 抗铁死亡亦未得到研究。上海交通大学乔永霞、于永春和王佳宜课题组联合探究了细胞外系统是否以及如何使 LUAD 细胞对铁死亡脱敏,并将成果发表在 Cancer communications(IF:15.283) 杂志上。

文献链接: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9017758 /
技术路线:


实验结果:
1. EV 拮抗 LUAD 中的脂质过氧化和铁死亡
从 LUAD 临床样本中发现 MDA 和 4 -HNE 水平要比正常组织低,而且 EGFR、KRAS 和 TP53 突变与 MDA 的关系不大,意味着在 LUAD 中脂质过氧化受到抑制。用 EV 抑制剂 GW4869 或 DMA 处理离体的 LUAD 后,4 -HNE 水平升高。因此作者用正常人和 LUAD 患者血浆 EV 处理 H1975 细胞 (对 erastin 和 RSL3 敏感),发现 LUAD-EV 能防止 erastin 和 RSL3 引起的 H1975 细胞活力下降、死亡及脂质 ROS 产生。A549 细胞 (对erastin和RSL3不敏感) 来源的 EV 或 DMEM 能使 H1975对erastin和RSL3脱敏,但会被 GW4869 预处理所抑制。说明 EV 拮抗 LUAD 细胞中脂质过氧化和铁死亡。

对 circRNA ChIP 数据库分析发现 cir93 和 cir34 在 LUAD 中表达上调,经过 IHC 和 QPCR 确认 cir93 在 LUAD 中高表达。基于肿瘤和其间质对肿瘤发展都很重要,因此通过 IHC 检测组织样本中 cir93,发现 cir93 主要表达于肿瘤区域,在成纤维细胞中不表达,另外在 EpCAM(+) 细胞中的表达水平明显高于 EpCAM(-) 细胞。虽 EpCAM(-) 和 EpCAM(+) 细胞释放的 EV 浓度无差异,但 EpCAM(-) 细胞培养基不能维持 EpCAM(+) 细胞中的 cir93 水平;GW4869 会降低 EpCAM(+) 细胞内 EV 浓度,但不影响 EpCAM(+) 细胞内 cir93 的含量,表明只有 EpCAM(+) 细胞的 EV 能维持 EpCAM(+) 细胞内 cir93。通过示踪实验进一步证实肿瘤细胞 EV 对提高 LUAD 肿瘤区域的 cir93 是必不可少的。
在小鼠身体两测分别注射带有不同荧光标记 cir93 的 A549 细胞,建模 18 天后分析发现两测肿瘤组织均可检测到荧光重叠信号,而这些重叠信号及组织内 cir93 的水平可被 GW4869 阻断。证实 LUAD 细胞在体内直接产生 cir93 并同时从 EV 中获得 cir93 以维持 cir93 的表达。

接着探究 EV 是否通过细胞内 cir93 来抗铁死亡。TEM 和 IF 分析发现在 H1975 细胞内过表达 cir93,可以抑制 erastin 和 RSL3 诱导的铁死亡;抑制 cir93, 则加剧 erastin和RSL3诱导的细胞死亡和 ROS 产生。同样,在 H1975 原位模型中过表达 cir93 可抑制铁死亡诱导剂 PKE 诱导的 4 -HNE 和 MDA 升高。使用 C11 -BODIPY 581 / 591 检测氧化脂质,确认 cri93 过表达能通过抑制脂质过氧化促使 LUAD 抗铁死亡。
花生四烯酸 AA 和肾上腺酸 AdA 的过度过氧化并掺入质膜是激活铁死亡的必不可少的步骤,但 H1975 细胞高表达 cir93 后会减少 AA 而非 AdA 掺入质膜。另外与成纤维细胞系来源的 EV 相比,A549 -EV 能显著降低 erastin 诱导的 ROS 生成。表明 EV 通过 cir93 抑制 LUAD 细胞中的脂质过氧化。

蛋白质组学和 RNA pulldown 分析出有 143 种蛋白质可与 cir93 相互作用,但经验证只有 FABP3 可被 cir93 上调。与 cir93 一样,FABP3 亦不受 erastin 和 RSL3 调节。此外在 FABP-/-细胞中,cir9 不能诱导 LUAD 抵抗 erastin 和 RSL3 的作用。
PAR-CLIP 确认了 FABP3 和 cir93 相互作用,catRAPID 预测了结合位点,通过 cri93 pulldown 和 RIP 实验分析,发现 FABP3 的第 96 - 102 位 aa 区域 (p#4) 与 cir93 的第 151st~202nd nt(R#2) 参与 cir93 -FABP3 相互作用。
细胞中过表达 FABP3 会降低 AA 掺入质膜中,但删除 p#4 或 R#2 后,不能阻止 AA 掺入质膜中,此外 cir93 -FABP3 相互作用是降低细胞内 AA 含量所必需的。破坏 cir93 -FABP3相互作用会消除 cir93 和 FABP3 促 H1975 细胞抗铁死亡能力。LUAD血浆EV能上调WT FABP3工程化H1975细胞中的FABP3,但对缺失p#4的工程化H1975细胞中FABP3表达基本无影响。表明cir93 -FABP3相互作用是EV上调FABP3需要,也是调节AA、降低脂质过氧化和LUAD细胞抗铁死亡所必需的。

5. 牛磺酸参与cir93上调FABP3以调节AA并使LUAD细胞抗铁死亡
对低表达和高表达FABP3的LUAD组织进行代谢组学分析,检测出牛磺酸是 wei 一一种可与AA反应生成NAT。通过ELISA和靶向MS分析,发现与突变的FABP3相比,WT FABP3在细胞内过表达后,减少了细胞内AA和牛磺酸;同时过表达cir93或用LUAD血浆EV处理细胞,也证实了cir93和LUAD-EV通过FABP3调节牛磺酸和AA。表明 FABP3抑制了AA掺入质膜,促进其与牛磺酸的反应,从而减少了细胞内总AA。
将牛磺酸合成底物CDO1和CSAD敲除后,细胞内牛磺酸降低,erastin和RSL3诱导的铁死亡和ROS产生明显增多,同时抑制了由cir93和FABP3介导的恢复作用。结合体内实验,证明牛磺酸是一种抗铁死亡因子,是 cir93 和 FABP3 减少 AA 所必需的。

6.NAT阻止AA掺入LUAD细胞质膜
过量外源 AA 存在情况下,虽cir93和FABP3抑制了AA掺入细胞膜,但也降低了细胞内AA的水平,推测NAT可能参与其中,通过细胞内过表达cir93和FABP3进行了验证,AA 消耗伴随 NAT 生成。LUAD-EV能进一步提高细胞NAT水平,说明NAT是EV使LUAD细胞抗铁死亡的关键因素。
将NAT直接与H1975孵育后,能抑制AA掺入质膜中,同时参与AA掺入质膜的酶ACSL4、LPCAT3和PLTP被NAT以剂量依赖性方式降低;NAT的存在抑制了erastin和RSL3诱导的细胞死亡和ROS产生。表明NAT通过抑制AA掺入质膜而诱导LUAD细胞对铁死亡脱敏。
生物信息学分析ACSL4、LPCAT3和PLTP基因的启动子,筛选出TLX2为3个基因表达的关键调控转率因子,且CHIP结果表明NAT通过抑制TLX2结合启动子来抑制ACSL4、LPCAT3和PLTP表达。TLX2过表达后能增强LUAD细胞抵抗erastin和RSL3的作用。表明TLX2在cir93 /FABP3 /NAT轴下游发挥调节LUAD抗铁死亡作用。

7.LUAD中EV和cir93、FABP3、牛磺酸及NAT之间的相关性
cir93 -/- A549细胞EV中基本检测不到cir93。正常A549细胞EV处理H1975细胞能调节ROS产生、FABP3、ACSL4、LPCAT3和PLTP的表达、牛磺酸、AA和NAT的浓度,但若敲除cir93,则EV不能发挥促细胞抗铁死亡作用。
在LUAD临床样本内也检测到FABP3高表达,同时与cir93具有显著相关性,牛磺酸与cir93和FABP3呈负相关,而NAT与cir93和FABP3呈正相关,LUAD血浆EV中cir93与细胞内cir93和FABP3呈正相关,因此在LUAD中EV和细胞内cir93、FABP3、牛磺酸及NAT之间的密切相关。
8.阻断EV能促进铁死亡
与具有高水平cir93和FABP3的PDX#2相比,PKE处理低表达cir93和FABP3的PDX#1时能诱导高水平的4 -HNE,离体组织切片进一步分析发现PDX#2对erastin诱导的铁死亡抵抗力更强,但该模型鼠总生存期要比PDX#1短。表明高水平cir93和FABP3促进肿瘤发展,诱导LUAD抗铁死亡。
用GW4869与PKE共处理A549的CDX模型,诱导了MDA的升高,显著抑制肿瘤生长,延长生存期。在细胞水平上,PKE处理A549细胞能抑制细胞增殖,GW4869或敲低cir93和FABP3,能进一步抑制细胞增殖,但这些作用可被铁死亡抑制剂Lipro- 1抑制。表明在LUAD中抑制EV/cir93 /FABP3后,铁死亡参与了肿瘤抑制,阻断EV或抑制其功能有助于改善基于铁死亡的疗法。

讨论:
本研究发现肿瘤细胞释放的EV增加细胞内cir93,以上调FABP3并使LUAD细胞通过FABP3依赖性方式降低总AA水平,诱导NAT产生,阻止AA掺入质膜中,从而引起细胞抗铁死亡。研究中作者提供了几个新的铁死亡靶点:cir93、FABP3、牛磺酸和NAT,鉴于它们的功能都与EV相关,因此通过阻断EV,可能有助于提高基于铁死亡的疗法在未来治疗LUAD中的疗效。